Urea Cycle
The urea or ornithine cycle is a series of biochemical reactions found primarily in the liver and, to some extent, in ureotelic animals such as amphibians and mammals. The pathway helps dispose of the highly toxic ammonia (NH3) from the blood by converting it to urea CO(NH2)2.
Ureotelic animals cannot excrete ammonia directly and convert it into a more straightforward form, the urea in the liver. The urea is then transported to the kidney through the bloodstream and excreted as urine.
Also known as the Krebs-Henseleit cycle, the urea cycle was discovered by Hans Krebs and Kurt Henseleit in 1932, a few years before the TCA cycle.
Steps of the Urea Cycle
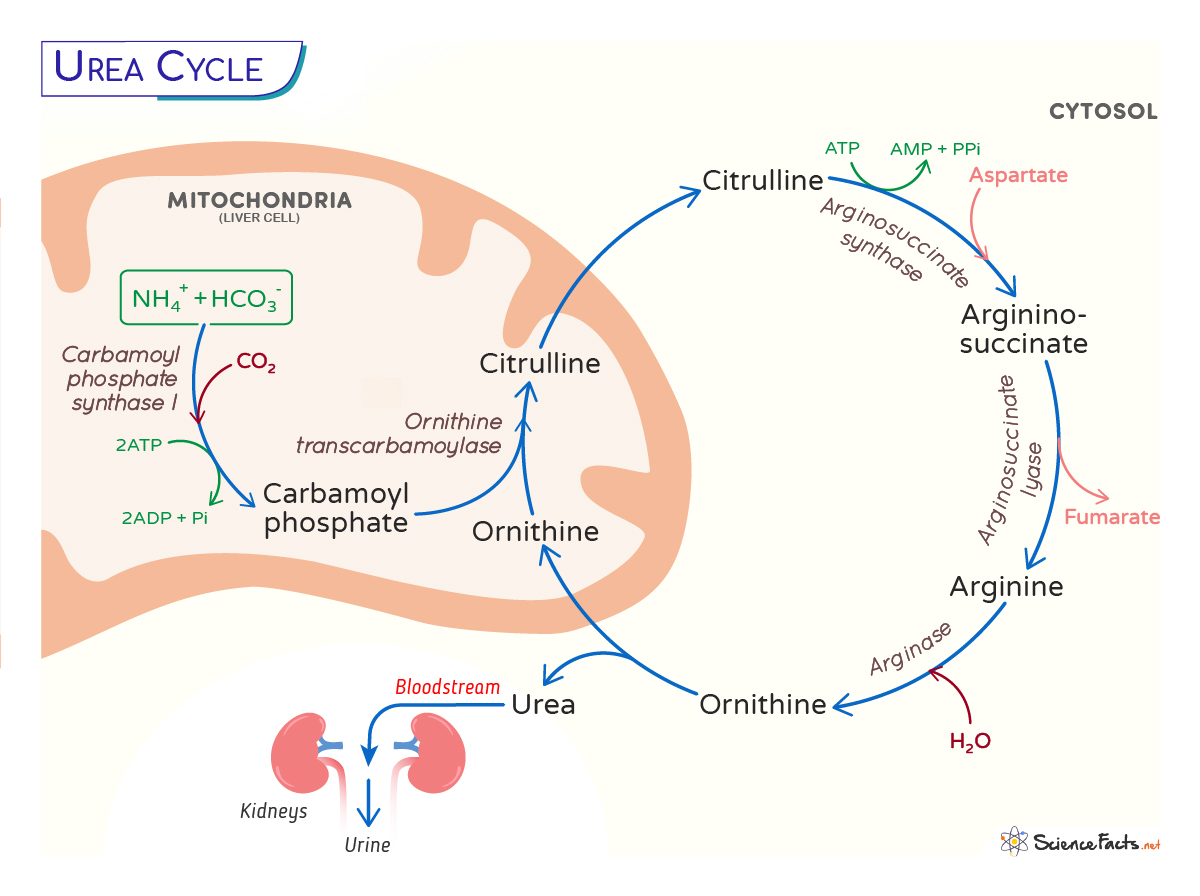
The urea cycle starts in the mitochondria of the liver cells and ends in its cytoplasm. There are five steps in the pathway. The first two steps occur in the mitochondria, the rest in the cytosol:
In the Mitochondria
Step 1: The first step involves entering ammonia into the cycle. Ammonia produced in the mitochondria makes carbamoyl phosphate by the enzyme carbamoyl phosphate synthetase I (CP I). The CPS1 requires an obligate activator, N-acetyl glutamate (NAG), and thus a rate-limiting step.
NH4+ + HCO3– + 2 ATP → Carbamoyl phosphate + 2ADP + Pi
It is a rate-limiting step.
Step 2: The second step involves transferring the carbamoyl group from carbamoyl phosphate to ornithine through the enzyme ornithine transcarbamoylase (OTC) to form citrulline.
Carbamoyl phosphate + Ornithine → Citrulline + Pi
In the Cytosol
The citrulline formed in the mitochondria is then released into the cytosol, where the rest of the reactions occur.
Step 3: In this step, the citrulline, with the help of an ATP molecule, forms an intermediate citrullyl-AMP intermediate, which further reacts with an amino group from aspartate to produce argininosuccinate. The enzyme arginosuccinate synthetase catalyzes this step.
Citrulline + ATP + Aspartate → Argininosuccinate + AMP + PPi
Step 4: The argininosuccinate is cleaved by the enzyme argininosuccinate lyase to form fumarate and arginine.
Argininosuccinate → Arginine + Fumarate
Step 5: The cycle’s last and fifth step involve hydrolysis of arginine from urea, catalyzed by arginase.
Arginine → CO(NH2)2 + Ornithine
The overall reaction of the urea cycle is given below:
NH3 + CO2 + aspartate + 3 ATP + 3 H2O → Urea + Fumarate + 2 ADP + 2 Pi + AMP + PPi + H2O
Regulation of the Urea Cycle
The regulation of the urea cycle depends mainly on two factors:
- Availability of Substrates: Except for arginase, all other enzymes of the urea cycle work based on the presence of the substrate. High ammonia levels will stimulate the urea cycle to eliminate excess nitrogen. In contrast, low levels may slow down the process.
- Allosteric Regulation: The enzyme CPS I, catalyzing the first step of the urea cycle, is allosterically regulated by NAG since it is an obligate activator of carbamoyl phosphate synthase.
NAG is generated when acetyl CoA and glutamate undergo a reaction catalyzed by NAG synthase, and this process is enhanced by the presence of arginine (Arg) and glutamic acid (Glu). Consequently, Glu serves as both a substrate and a catalyst for the urea cycle.
Besides the above two factors, the urea cycle is also regulated by the transcriptional and translational modification of the enzymes. Hormones like insulin stimulate the production of the amino acid metabolites that can enter the cycle. In contrast, hormones glucagon and cortisol promote the breakdown of proteins, leading to increased ammonia production, thus activating the urea cycle.
What is the Function of the Urea Cycle
Accumulation of excess urea in our body is toxic. The primary purpose of the urea cycle is to eliminate this excess ammonia from the body.
A healthy adult human being removes about 10 to 20 g of ammonia daily from their body. The accumulation of these nitrogenous compounds will lead to urea cycle disorders. In most cases, such disorders are caused by a deficiency of one or more cycle enzymes.
Disorders of the Urea Cycle
Some of the common disorders caused by defects in urea metabolism are actually due to the defects in the enzymes catalyzing a particular step. These disorders are:
- Carbamoyl phosphate synthetase I (CPS I) deficiency
- Ornithine transcarbamylase (OTC) deficiency
- Argininosuccinate synthetase deficiency
-
References
Article was last reviewed on Friday, October 20, 2023
Related articles
Popular Articles
Join our Newsletter
Fill your E-mail Address